
About Us
About Us
ACS Catal.: “从头”预测纳米材料模拟过氧化物酶活性
近日,ACS Catalysis期刊上线了江西师范大学高兴发教授和国家纳米科学中心赵宇亮院士团队的合作研究论文“Density Functional Theory-Based Method to Predict the Activities of Nanomaterials as Peroxidase Mimics”,这个工作基于化学反应原理和密度泛函计算提出了一个更具普适性的预测POD纳米酶活性大小的理论方法。
○ 研究背景 ○
目前实验已报导了300余种过氧化物酶(POD)纳米酶,它们与天然POD类似,可催化如图一所示的显色反应,可替代POD在生物传感、抗菌和有机污染物降解等应用领域发挥作用。
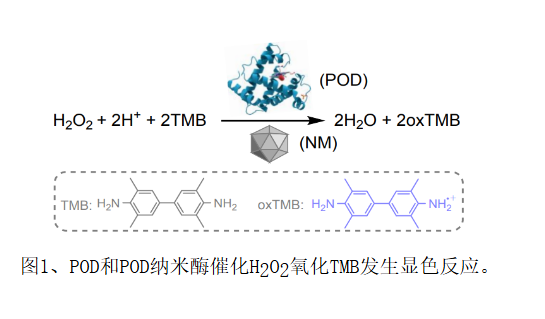
然而人们对POD纳米酶催化反应动力学的原子水平认识并不深刻,缺乏预测POD纳米酶活性的普适性方法。例如,之前计算研究发现,H2O2在贵金属Au、Ag、Pd和Pt表面的分解能垒和吸附能可以预测贵金属的POD活性,然而此预测方法并不适用于还原性更强的其他金属。对于四氧化三铁Fe3O4这类同时包含二价铁Fe2+和三价铁Fe3+的金属氧化物,有研究发现Fe2+含量升高,POD活性增强。但Fe2+含量和POD活性间的正相关关系不可推广,如氧化亚铁FeO中只含有Fe2+,但其POD活性却显著低于Fe3O4。
发展更加普适的理论方法预测POD纳米酶活性对纳米酶的理性设计和筛选具有重要意义。Fe3O4纳米颗粒是研究最早、目前最受关注的POD纳米酶之一,因此本研究采用DFT计算方法研究氧化铁表面模拟POD催化的机理和动力学,以期导出预测POD纳米酶活性的普适性理论方法。
○ 研究内容 ○
本研究构造了如图2所示的15种纳米氧化铁表面,它们分别衍生自氧化亚铁(FeO)、四氧化三铁(Fe3O4)或三氧化二铁(Fe2O3)晶体,具有不同的晶格结构、暴露晶面、结构缺陷或表面修饰,因此含有的Fe2+/Fe3+比例及表面Fe2+的分布状况均不同,为氧化铁类酶活性的计算和模拟提供了较完备的模型。
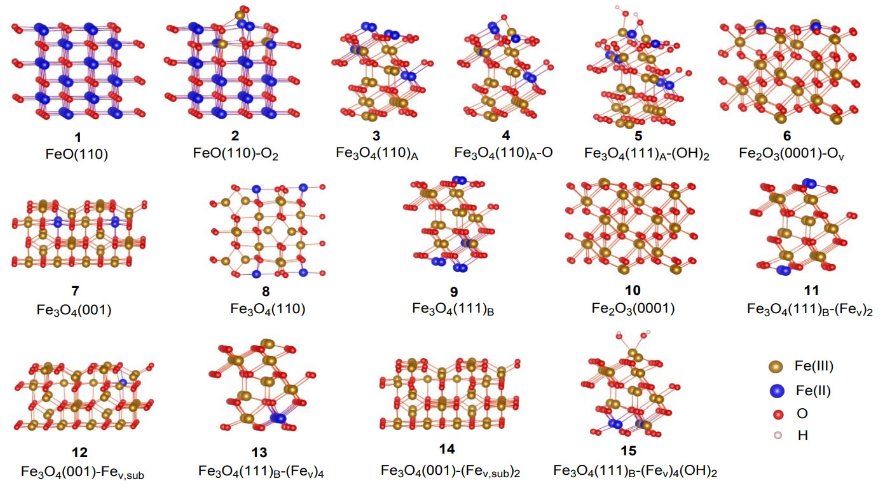
图2、不同晶体结构、暴露晶面、结构缺陷和表面修饰的氧化铁表面。
计算表明该15种氧化铁表面遵循相似的类POD催化机理(图3a)。首先,H2O2分子在纳米材料表面吸附并分解产生2个OH吸附结构(step 1:i → iii)。对于一些还原性强的氧化铁表面(表面Fe2+离子丰富的表面),该反应步无能垒;对于还原性弱的氧化铁表面(表面Fe3+离子丰富的表面),该步反应会经历一个过渡态结构ii‡并伴随能垒Eb,1。接下来的主要反应是中间体结构iii中的两个OH基团陆续被TMB还原,即step 2(iii → iv‡ → v)和step 3(v → vi‡ → i),它们伴随各自的反应能垒Eb,2和Eb,3。该催化机理与已知的贵金属、钙钛矿以及碳材料表现POD活性的机理相似。
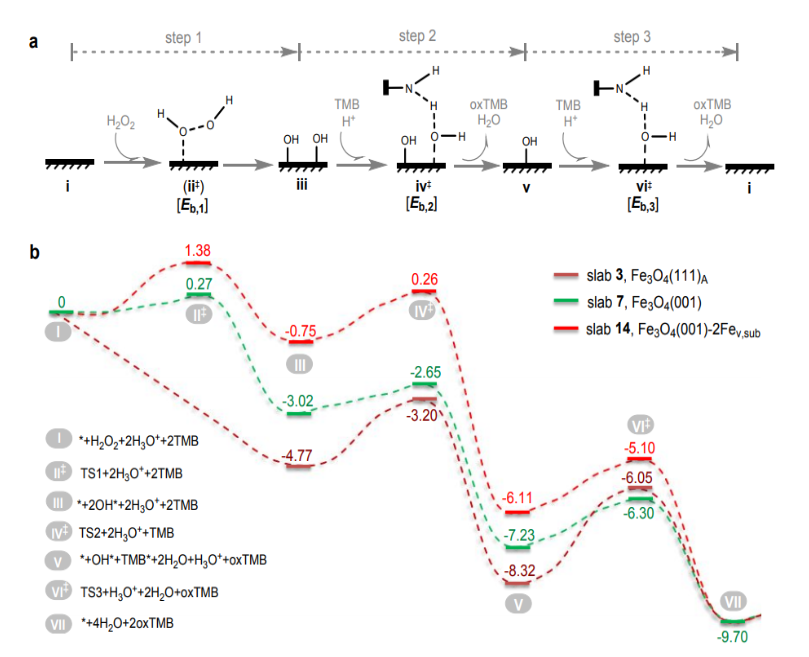
图3、DFT计算得到氧化铁表面表现POD活性的催化反应机理和动力学。(a)氧化铁表面POD活性分子机制。(b) 氧化铁表面3、7和14的催化反应能量曲线(能量单位:eV)。
虽然15种氧化铁表现POD活性时遵循相似的催化机制,但是反应动力学却不一样:决速步可在step 1和step 3之间变换(图3b)。因此step 1和step 3的反应能和能垒(Er,1, Eb,1, Er,3和Eb,3)决定整个催化反应的效率。基于化学反应原理进行近似处理和推导,可知Er,1, Eb,1, Er,3和Eb,3均和Er,1之间有线性关系,因此Er,1是描述该催化活性的理想描述符。由于Er,1值与该表面对羟基自由基的吸附能Eads,HO间也有线性关系,因此Eads,HO也是氧化铁表面POD活性的理想描述符。以Eb,RDS < 1.5 eV为预测氧化铁表面具有POD活性的判据,可以得到如下结论:当氧化铁表明满足条件-3.6 eV < Er,1 < -0.3 eV(或-3.5 eV < Eads,OH < -1.6 eV)时具有POD活性,当Er,1 = -2.1 eV(或Eads,OH = -2.6 eV)时POD活性最强。
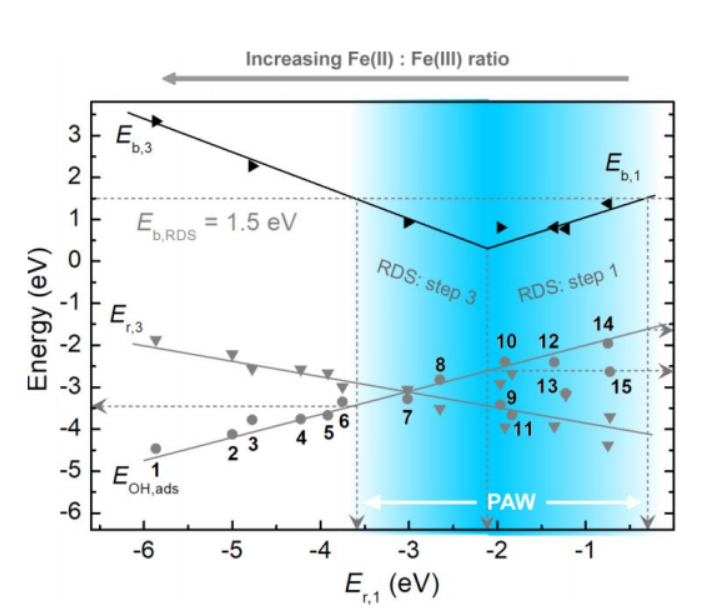
图4、氧化铁表面POD活性的火山型曲线。
为检验上述预测模型的可靠性,除15种氧化铁表面外,另计算了共计57种金属、单金属氧化物、钙钛矿和碳材料的Eads,OH值(图5)。结果表明,经实验证实的POD纳米酶的Eads,OH均落在POD活性窗口(PAW)内,这表明预测模型确实具有可靠预测能力,同时也表明图3a所示的催化机理对不同种类POD纳米酶具有普适性。
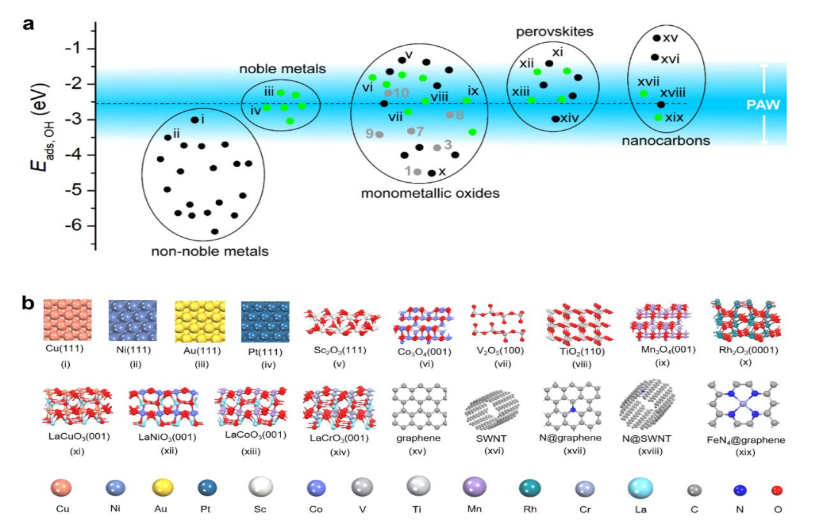
图5、纳米材料POD活性预测。(a)蓝色区域为通过Eads,OH定义的POD活性窗口(PAW),蓝色到白色的渐变代表活性从高到低的转变;绿色圆圈标出的纳米材料的POD活性已被实验报道,灰色圆圈代表本文中研究的氧化铁表面。(b) a中用罗马数字标出的纳米材料结构图。
6、结论与展望
本研究通过DFT计算研究了氧化铁表面发挥类似于POD催化功能的分子机理和动力学,提出了预测POD纳米酶活性的理论方法。该方法不仅适用于氧化铁,也适用于其他具有相似POD催化机理的纳米材料,预测结果与目前已知的POD纳米酶实验结果吻合。因此,它可望发展成为一种通用而稳健的方法,用于计算机辅助POD纳米酶筛选和设计。
原文:
Shen, X.; Wang, Z.; Gao, X.;* Zhao, Y. Density Functional Theory-Based Method to Predict the Activities of Nanomaterials as Peroxidase Mimics, ACS Catal. 2020, DOI: 10.1021/acscatal.0c03426.
原文链接:
https://pubs.acs.org/doi/10.1021/acscatal.0c03426