
第一作者:Yang Gao
通讯作者:薛玉瑞、黄长水、李玉良
通讯单位:中国科学院化学研究所、中国科学院大学
研究背景
1.海水电解面临的挑战:海水电解是可持续氢能转换的有前景方向,但其效率受限于阳极的选择性低、反应动力学差及电极腐蚀等问题。现有催化剂效率远低于天然系统。
2.单原子催化剂的优势与挑战:单原子催化剂因其特定物理化学性质在催化和材料科学中受到重视,但精确控制金属原子的锚定仍具挑战,包括复杂的合成过程和金属原子的易聚集。
3.GDY的独特性能与应用前景:GDY的结构和性能满足新能源材料的需求,特别是其丰富的炔基孔和高还原性使其成为单金属原子锚定的理想平台,克服传统催化剂的缺陷并提升电解器性能。
研究内容
3.优越的氧析出反应性能:该设计系统在300 mV低过电位下表现出前所未有的OER性能,质量活性达2.6 A mgcat.-1,是IrO2催化剂的216.6倍,且具有长期稳定性。
图文解析
要点1:高度有序钴-铱原子阵列的生长过程
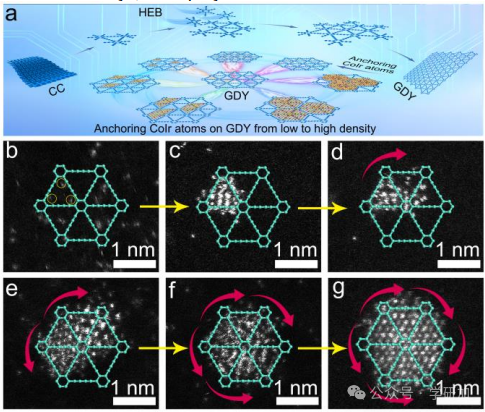
图1 Co1Ir3原子阵列在GDY上的逐步锚定过程。
本文介绍了在全晶体GDY上有序生长钴-铱原子的方法,通过逐步锚定金属原子,在常温下实现了高效选择性生长。
图1a:展示了通过直接控制逐步锚定金属原子,在常温下在2D GDY上有序生长钴-铱原子的过程。该过程利用了GDY中丰富的sp-C杂化三键骨架、
空间限制效应、不完全电荷转移和乙炔键的高还原性,使金属原子实现选择性和受控生长。
图1b:使用HAADF-STEM表征了在GDY表面锚定的单个金属原子,显示了低密度金属负载情况下的单个钴和铱原子分布,验证了初始金属原子锚定的成功。
图1c:进一步展示了单个金属原子作为成核位点,额外金属
原子在其周围锚定的过程,表明成核位点的金属原子能有效吸引并固定更多的金属原子。
图1d:随着反应时间的延长,金属原子从一个三角腔逐步锚定到另一个三角腔,展示了金属原子在不同位置的连续锚定过程,说明了时间对金属原子生长位置的影响。
图1e:展示了金属原子在不同三角腔内连续锚定的进一步过程,形成更复杂的金属原子阵列结构。
图1f:反应时间进一步延长后,形成了具有明确金属原子阵列的六边形结构,展示了高度有序的金属原子排列。
图1g:展示了高密度金属原子阵列在GDY上的生长,显示出高度有序的金属原子排列和高度密集的阵列结构,验证了GDY在高密度金属原子锚定中的优势。
小结:通过逐步锚定和控制反应时间,在GDY上实现了从低到高密度金属原子阵列的有序生长,验证了GDY在金属原子锚定中的优越性能。通过Raman光谱证实了GDY在合成过程中的结构完整性。
要点2:GDY纳米片的结构和金属原子锚定分析

图2 Co1Ir3/GDY的结构与元素分布分析。
本文通过多种表征手段展示了GDY纳米片的3D多孔结构及其高结晶度,以及钴-铱原子在GDY表面的均匀分布和高质量负载。
图2a:SEM图像展示了GDY纳米片在碳纤维表面的垂直排列和互联,形成了3D多孔结构。
图2b和2c:HRTEM图像显示了GDY的高结晶度,图中可见晶格条纹,证实了GDY的晶体结构。
图2d:SAED图像进一步确认了GDY的高结晶度,显示出清晰的衍射点,支持晶格条纹的观察结果。
图2e和2f:HRTEM图像显示了0.46 nm的晶格条纹和ABC堆积模式,验证了GDY的结晶特性。
图2g和2k:展示了GDY纳米片在锚定钴/铱原子后的形态和结晶性质未受到显著影响,保持了原有的结构。
图2h和2i:显示了钴-铱金属原子阵列在GDY表面的均匀分布,形成了六边形结构。
图2j:展示了由于钴-铱原子的锚定,GDY的晶格间距增加到0.47 nm。
图2l-2o:元素映射结果显示了Ir、Co和C在Co1Ir3/GDY纳米片上的均匀分布,验证了金属原子的均匀锚定。
小结:通过SEM、HRTEM、SAED和XRD等多种表征手段,证实了GDY纳米片的高结晶度和多孔结构,并展示了钴-铱原子在GDY表面的均匀分布和高质量负载,保持了原有的晶体结构。
要点3:电子结构和催化性能分析
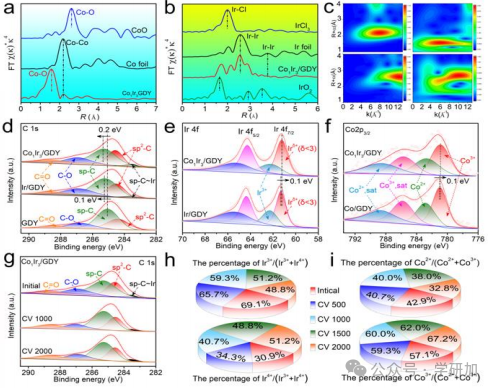
图3 钴铱催化剂的电子结构与循环稳定性分析。
本文通过X射线吸收光谱和X射线光电子能谱测定复合材料的电子结构,探讨Ir和Co在催化过程中对性能的影响。
图3a:在Co-K边的κ3加权EXAFS傅里叶变换谱显示出一个显著峰值在约1.6 Å,及在约2.2 Å处非常弱的峰值,表明Co在CoIr/GDY中原子分散。
图3b:在Ir-L3边的κ3加权EXAFS显示出约2.5 Å的显著峰值,表明Ir-Ir/Co原子间的相互作用。
图3c:Co和Ir EXAFS振荡的波谱变换(WT)分析,Ir箔和IrCl3显示最大强度分别在约12 Å−1和7.8 Å−1,而Co1Ir3/GDY在约9 Å−1,指示Ir-Ir/Co贡献。
图3d:C 1s XPS数据表明Co1Ir3/GDY和Ir/GDY之间形成了强sp-C–金属键。
图3e:Ir 4f XPS数据显示,与对照样品相比,Co1Ir3/GDY的sp–C峰正移0.2 eV,Ir 4f峰负移0.1 eV。
图3f:Co 2p3/2 XPS数据与对照样品相比,显示负移0.1 eV,指示GDY与金属原子之间的不完全电荷转移。
图3g:C 1s XPS数据显示在2000次循环测试后没有能量变化,表明GDY结构的稳定性。
图3h:Ir 4f XPS数据显示,随着OER电催化过程进行,Irδ+(δ<3)减少,Ir3+增加。
图3i:Co 2p XPS数据表明,随着循环测试增加,Co2+减少,Co3+增加。
小结:通过X射线吸收和X射线光电子能谱分析,揭示了Co和Ir在催化过程中保持混合价态的重要性,这有助于提高和维持催化活性。
要点4:Co1Ir3/GDY催化剂在海水裂解中的活性和稳定性
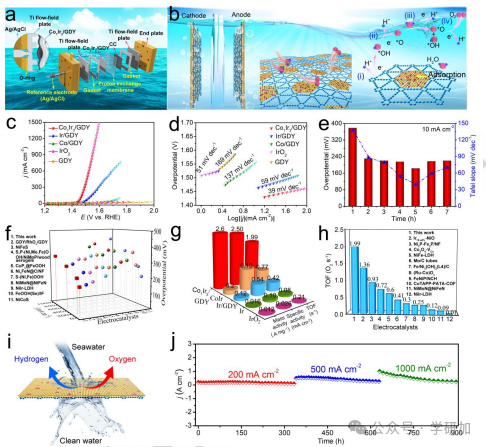
图4 钴铱催化剂的高效氧气生产和长期稳定性测试。
图4评估了Co1Ir3/GDY在碱性模拟海水中的催化活性和稳定性,通过电解槽将阳极和阴极分开,以防止氢气和氧气混合。
图4a和4b:展示了使用质子交换膜将阳极和阴极分开的电解槽装置,以防止氢气和氧气混合。
图4c:线性扫描伏安曲线(LSV)显示,高密度Co1Ir3/GDY在200、500、1000和1500 mA cm-2电流密度下的过电位分别为246、282、325和366 mV,远低于商用IrO2等参考催化剂。
图4d:Tafel斜率图显示,Co1Ir3/GDY的Tafel斜率为39 mV dec-1,表明其OER动力学最快。
图4e和4f:与商用IrO2及其他报道的OER催化剂相比,Co1Ir3/GDY在各种电流密度下的过电位更低,表明其具有卓越的催化性能。
图4g:质量活性、比活性和周转频率(TOF)表明,Co1Ir3/GDY的质量活性为2.6 A mgcat.-1,比活性为2.50 mA cm-2,TOF为1.99 O2 s-1,显著高于商用IrO2及其他催化剂。
图4i和4j:长期稳定性测试显示,Co1Ir3/GDY在实际应用条件下工作905小时,在200、500和1000 mA cm-2电流密度下未出现故障。
小结:Co1Ir3/GDY在碱性模拟海水中展现出高活性和高选择性的OER性能,具有低过电位、高质量活性和长时间的稳定性。扫描电子显微镜(SEM)和HAADF-STEM图像显示,在稳定性测试后催化剂的形貌和化学结构保持完好。
总结展望
综上所述,研究者展示了一种简便的方法,通过逐步可控地在GDY表面锚定钴和铱原子,获得高密度Co1Ir3/GDY金属原子催化剂。
高密度的金属原子均匀分布在GDY表面,大大提高了质量活性和比活性。GDY表面电荷的不均匀分布以及GDY与金属原子之间独特的不完全电荷转移,产生了大量暴露的活性位点和高本征催化活性。
Co1Ir3/GDY展示了高达2.6 A mgcat.-1的质量活性,是现有最先进催化剂的216.6倍,并且在905小时的电解过程中,分别在200、500和1000 mA cm-2的电流密度下保持稳定和高效。
这项工作朝着设计具有预定结构的原子精确活性中心以用于电催化迈出了重要一步。
https://onlinelibrary.wiley.com/doi/epdf/10.1002/anie.202406043

